









This article looks at photocatalytic water oxidation and producing an artificial water photooxidation system, including how this could be used to generate renewable energy. It will help you understand the research the journal article is based on, and how to read and understand journal articles. The research article was orginaly published in our Chemical Science journal.
Photocatalytic water oxidation at soft interfaces
Click here to view the full article in the journal Chemical Science
Click here to view the article in Chemistry World
Authors: Malte Hansen, Fei Li, Licheng Sun and Burkhard König
Why is this study important?
The search for alternative renewable energy resources is one of the biggest scientific challenges of our times. 1, 2 Water as an abundant resource would be the ideal source for the production of the sustainable energy carrier hydrogen. Utilizing solar radiation as an inexhaustible energy source could drive the energetically uphill water splitting reaction. To find suitable catalyst systems (further resources on catalysis here and here) for photochemical water splitting into oxygen and hydrogen the overall reaction is divided into the reductive and oxidative half reaction, respectively. The water oxidation half reaction involving four electron transfer steps and highly reactive intermediates is considered particularly difficult. 3
A typical photochemical water oxidation system consists of two subunits: a light absorbing photosensitizer or dye and the water oxidation catalyst. The two subunits can be covalently connected which ensures an efficient electron transfer (electron transfer is distance dependent), but requires the synthesis of complex ligands and linkers. 4-8 If dye and oxidation catalyst are prepared as separate entities different combinations can be easily realized, but the electron transfer between the subunits in homogeneous solution is diffusion controlled and depends on the concentration. 9
Further information
- A review paper on ‘Surface tuning for oxide-based nanomaterials as efficient photocatalysts’
- Selected further reading from RSC books:
- Molecular Solar Fuels
- Photoelectrochemical Water Splitting
- Solar Energy Conversion
- Solar Hydrogen
What is the objective?
To create a working photochemical water oxidation system which combines the advantages of covalently bound assemblies and homogeneous systems by embedding the two redox partners (further resources on redox here and here) into a phospholipid bilayer membrane. The co-embedding ensures a close proximity and high local concentrations of the subunits and thus facilitates the electron transfer while it still allows the easy variation of photosensitizer-catalyst combinations, ratios and concentrations.
What was their overall plan?
- Choose an appropriate literature known homogeneous photochemical water oxidation system.
- Modify these compounds for the introduction into a phospholipid bilayer.
- Co-embed the amphiphilic derivatives into small unilamellar vesicles and perform photochemical water oxidation.
- Compare the vesicular system with the homogeneous one under identical conditions.
- Test the versatility of the method by embedding of different water oxidation catalysts.
- Investigate the influence of the bilayer membrane on the catalytic performance.
What was their procedure?
Choose an appropriate literature known homogeneous photochemical water oxidation system
The authors decided to choose a photochemical water oxidation system consisting of 2b a derivative of the well-known photosensitizer Ru(bpy)3 and 6a a ruthenium complex bearing a ligand with two carboxylate functionalities which was originally published by the Sun group. 10 The complexes were fully characterized by NMR, MS and X-Ray and the system produced oxygen under irradiation when sodium persulfate was used as a sacrificial electron acceptor. The photooxidation of Ru(bpy)3 and its derivatives with persulfate is an established and intensively investigated reaction. 11 The generated oxidized photosensitizer-species has a sufficient oxidation potential to oxidize the catalyst.
Modify these compounds for the introduction into a phospholipid bilayer
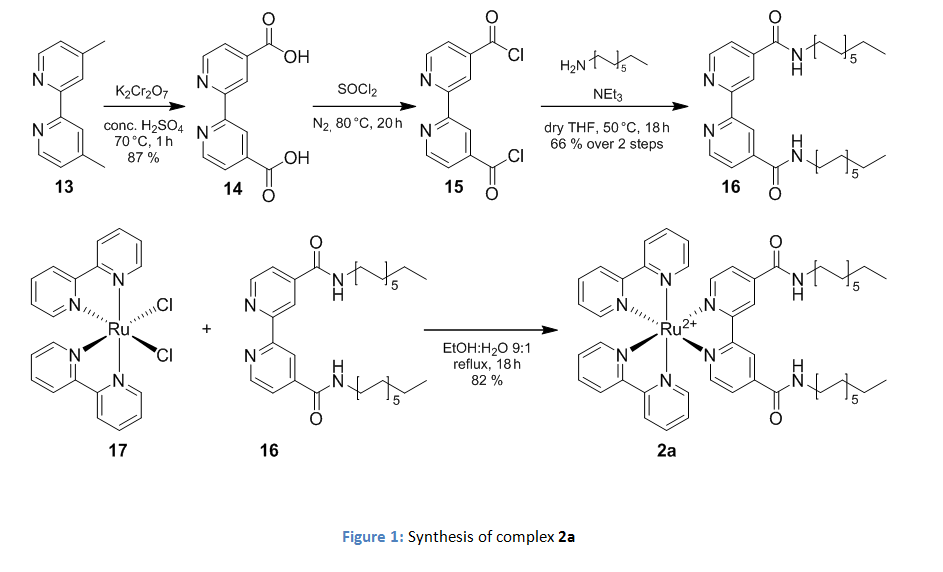
The necessary amphiphilicitiy was created by the introduction of alkyl-chains. In case of the photosensitizer 2b the amphiphilic derivative was synthesized with two dodecyl amide moieties instead of the ethyl esters as described in figure 1.
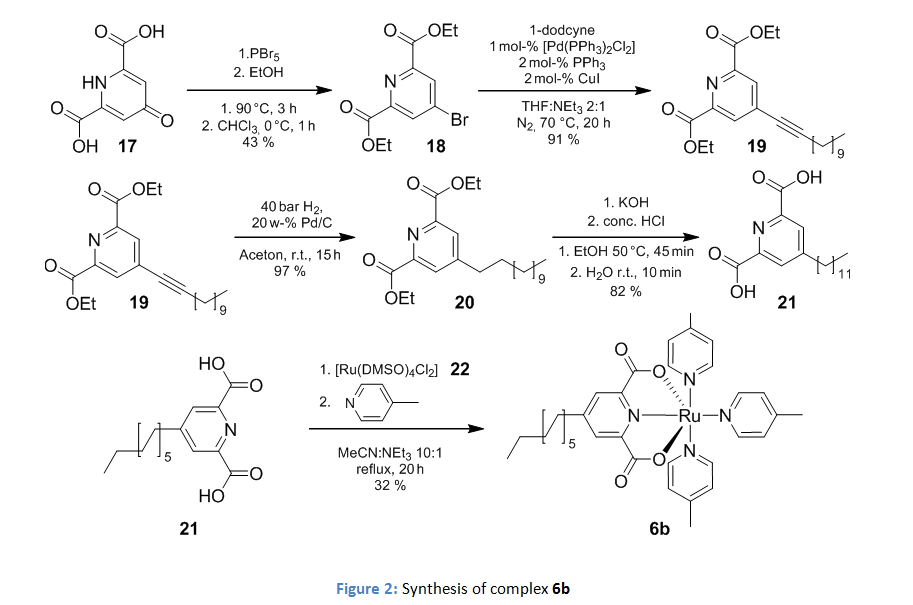
The catalyst was modified by the introduction of a dodecyl chain on the 4th position of the dipicolinic acid as shown in figure 2.
Co-embed the amphiphilic derivatives into small unilamellar vesicles and perform photochemical water oxidation
Functionalized vesicles were prepared by mixing appropriate volumes of stock solutions of 2a, 6b and a phospholipid (figure 5) in organic solvents in a crimp-top vial. After evaporation of the solvent phosphate buffer containing sodium persulfate was added and the vial was sonicated to obtain small unilamellar vesicles (figure 3). The size distribution of the vesicles was determined with dynamic light scattering and the sample was degassed with argon. After irradiation with blue LEDs the amount of evolved oxygen was measured by gas chromatography (figure 4).
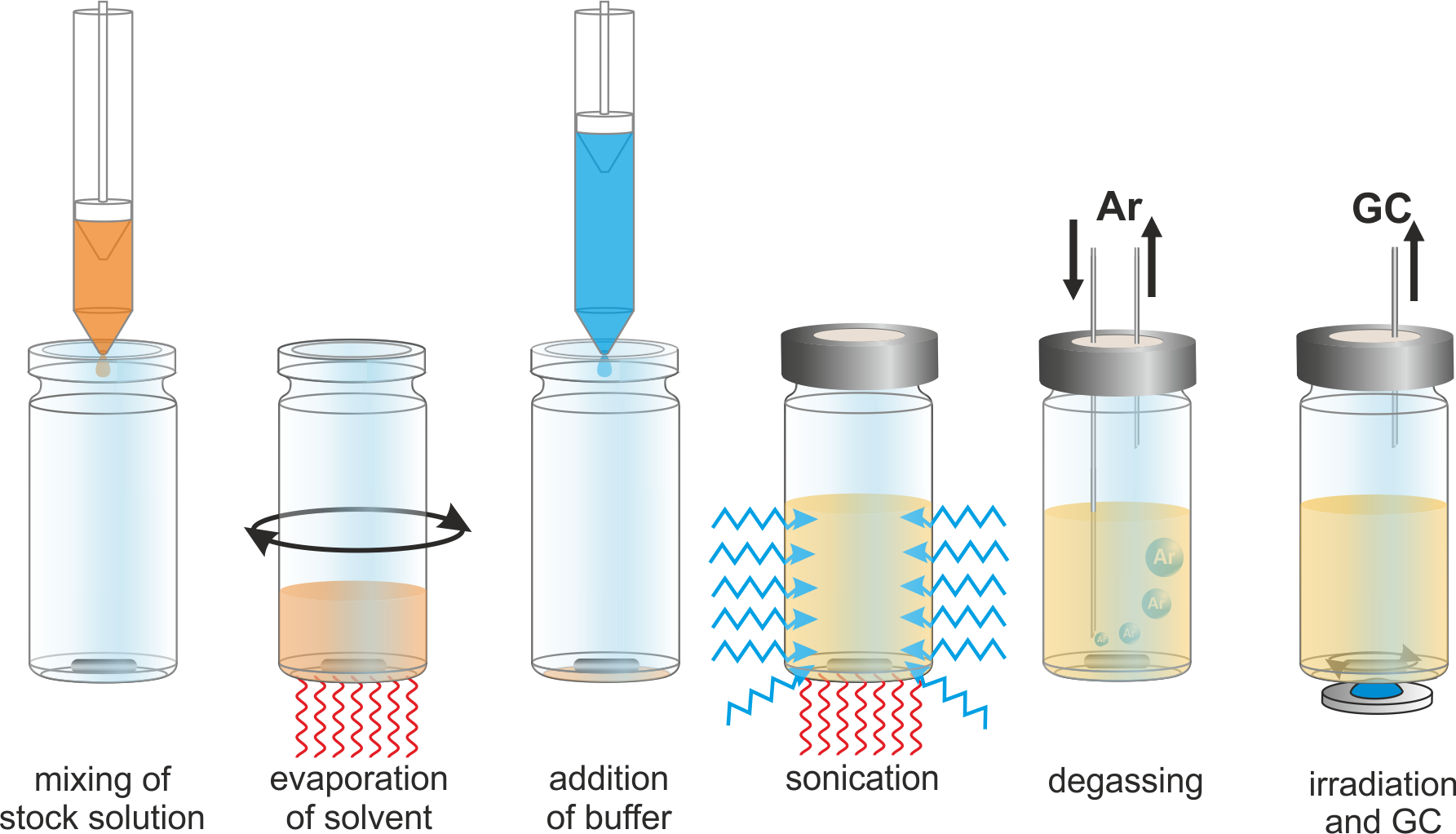
Figure 3: Schematic representation of the vesicle preparation.

Figure 4: Image of the LED irradiation and cooling device (left), schematic representation.
Compare the vesicular system with the homogeneous one under identical conditions
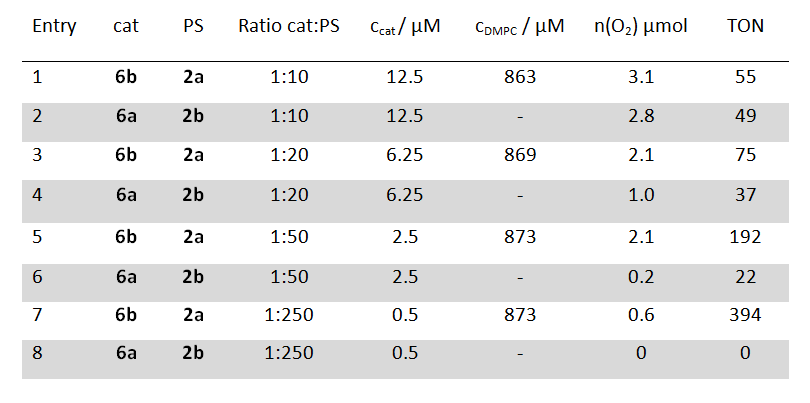
A comparison of the catalytic performance between homogeneous and vesicular systems under identical overall concentrations of photosensitizer and catalyst gave comparable turnover numbers. However, when the concentration of the catalyst was lowered the homogeneous system became less efficient, but the turnover number (TON) of the catalyst in the vesicular system increased significantly (table 1). This can be explained by the two dimensional arrangement of the two subunits in the membrane. The redox partners stay in close proximity, which facilitates the electron transfer, even at very low overall catalyst concentrations. This makes water oxidation possible at concentrations which cannot be realized in homogeneous systems.
Table 1: Evolved molecular oxygen after 20 min light irradiation and TON of photosensitizers (PS) 2 (125 µM) and water oxidations catalysts (cat) 6 in homogeneous solution (shaded in grey) and DMPC phospholipid vesicles at identical concentrations and a sodium persulfate concentration of 2.5 mM.
Test the versatility of the method by embedding of different water oxidation catalysts
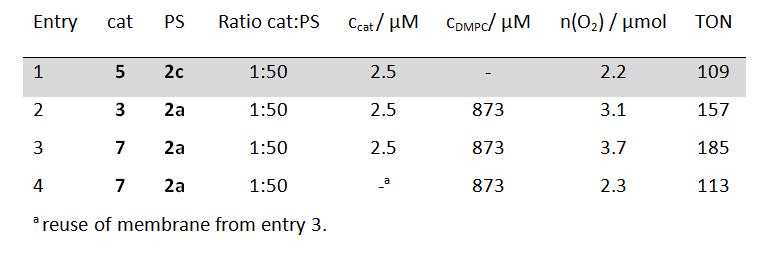
To prove the versatility of embedding catalyst-photosensitizer combinations into phospholipid bilayers the authors modified two other literature known photochemical water oxidation catalysts with alkyl chains. 12, 13 Vesicles functionalized with these catalysts also showed better catalytic activities than a homogeneous system under otherwise identical conditions (table 2). The literature known fact that the photosensitizer is the main limiting factor in homogeneous catalysis of these systems was also confirmed in the vesicular solutions, by the addition of new photosensitizer to used vesicles. These renewed vesicular solutions regained 60 % of the initial catalytic performance.
Table 2: Evolved molecular oxygen after 20 min light irradiation and TON of photosensitizers (PS) 2 (125 µM) and water oxidations catalysts (cat) 3 and 7 embedded in DMPC phospholipid vesicles; for comparison the performance of a homogeneous solution of catalyst 5 and photosensitizer 2c at identical concentrations is given (shaded grey). Sodium persulfate concentration: 2.5 mM.
Investigate the influence of the bilayer membrane on the catalytic performance
The physical properties of phospholipid membranes change drastically with the nature of the lipid. To investigate the effect on water oxidation activity vesicles with three different lipids were prepared. The three lipids have the same dipolar head group and differ only in the length and structure of the hydrocarbon chains (figure 5).
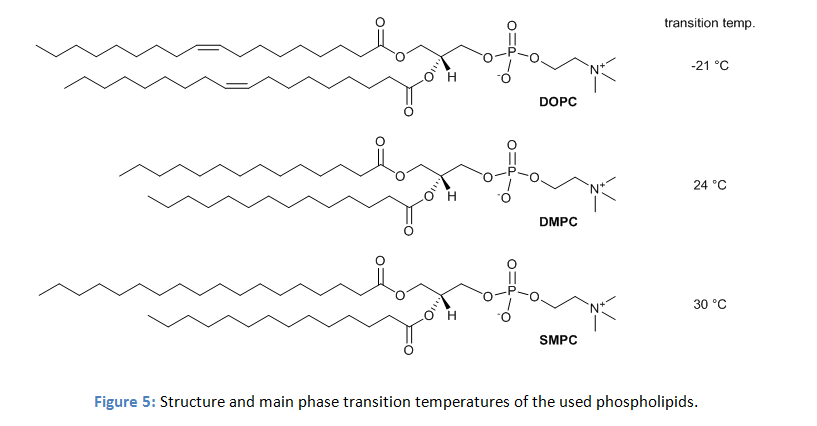
However, in DMPC and SMPC the co-embedded complexes produced oxygen upon irradiation, while the activity in DOPC was significantly lower. The authors explain the effect by the distinct differences in membrane fluidity. Phospholipid membranes show a significant change in the membrane fluidity at distinct temperatures. This is the transition from the relatively rigid gel phase to the liquid crystalline phase with high lateral diffusion. DMPC and SMPC have main phase transition temperatures from the gel to the liquid crystalline phase of 24 °C and 30 °C, but DOPC with a transition temperature of -21 °C is already in the liquid crystalline phase at the temperature of the experiment. Photooxidation experiments of DMPC and SMPC vesicles with embedded photosensitizers and catalyst at temperatures above their transient temperatures showed a reduced activity, while the activity remains unchanged at temperatures below the phase transition.
Table 3: Dependence of evolved molecular oxygen after 20 min light irradiation and TON of co-embedded photosensitizer 2a (125 µM) and water oxidations catalyst 6b (12.5 µM) depending on the phospholipid and the reaction temperature at a sodium persulfate concentration of 2.5 mM.
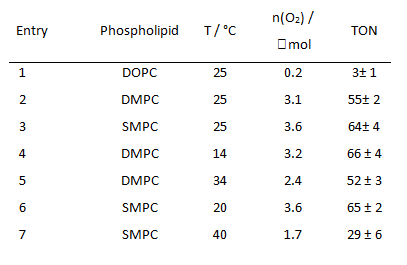
The authors conclude that phase separation and clustering of the embedded complexes, expected for the amphiphilic additives below the transition temperature of a lipid membrane, enhance the photocatalytic activity of the assembly.
What are the conclusions?
In summary, the authors have, for the first time, self-assembled photosensitizer - catalyst water oxidation systems by co-embedding two amphiphilic ruthenium complexes into the phospholipid bilayer membrane of small unilamellar vesicles. The observed oxygen production upon light irradiation is comparable to similar systems in homogeneous solutions, but superior at low concentrations of the water oxidation catalysts. Membrane embedded water photooxidation systems remain catalytically active at concentrations, where homogeneous mixtures of photosensitizer and water oxidation catalyst are inoperable. Phase separation and patch formation cluster the complexes in the membrane, which might facilitate the intermolecular electron transfer processes. The fluidity of the membrane affects the self-organization of the embedded complexes and therefore their photocatalytic performance. Highest TONs are observed in gel phase membranes, where phase separation is favoured. The method was applied to different combinations of sensitizers and oxidation catalysts and allows a rapid screening of sensitizer – catalyst combinations, ratios and concentration ranges.
What are the next steps?
Functionalized vesicles could provide a new approach for the creation of a water splitting device when combined with a hydrogen evolution electrode. This could be realized as a wired device with immobilized membranes on cathode and anode or as a Z-scheme water splitting system utilizing a redox mediator.
Professor Burkhard Koenig, one of the authors of the article, answers some additional questions on his group’s work.
Explain the focus and findings of your article and why it is of current interest?
Catalytic water splitting by visible light is one of the great challenges of our times, as it is closely connected with the sustainable energy production of the future. Typical catalytic systems work either in solution (homogeneous) or as solid (heterogeneous), but natural photosynthesis, which may serve as a model, uses a different approach: dyes and redox enzymes (biocatalysts) bound to fluid membranes. The key difference of membrane-bound processes to solution phase reactions is going from three dimensions to two dimensions, which has drastic implications on the concentration dependence of bimolecular processes. The difference to solid (heterogeneous) catalysts is that they are static, while membranes are fluid and dynamic; they can adapt and rearrange during reactions, which may enable processes like self-repair.
What distinguishes your work from other photosystems in bilayer membranes that have been reported in recent years?
We report the first example of a water photooxidazing system that is membrane bound. Homogeneous and heterogeneous catalysts for water oxidation have been reported. The key difference of the membrane bound system is that it still works at very low concentrations, where homogeneous systems fail.
What was the motivation behind the research?
I like to use inspirations from nature to design functional chemical systems.
How big an impact do you see your work potentially having?
At the current stage of development this is (like always) difficult to foresee.
Which part of you work proved the most challenging?
Optimizing the conditions and establishing reaction conditions that ensure good reproducibility of the water oxidation performance.
Could you expand more on the applications you see the membranes being employed in, considering the amount of oxygen they are able to generate?
Regarding applications in functional devices for artificial photosynthesis, two approaches may be considered:
1. To combine the water oxidation unit with the hydrogen generation unit in solution, and to use a redox shuttle for a soft “device” pretty much like the Z-scheme of PSII and PSI;
2. To immobilize the two units on the respective anode and cathode for a wired device.
The concentration/stability of the photosensitiser is identified as being the limiting factor and you report a 60% recovery of initial TON when a new photosensitiser is embedded into the membrane. How does this compare to the more traditional homogeneous systems that have been reported and is the reuse of your vesicles more complicated to achieve?
The stability of the photosensitizer is indeed a limitation in all reported systems. This limitation remains also in the membrane bound catalysts. Regeneration of catalyst is simple (addition of new amphiphilic sensitizer) and the recovery is comparable to homogeneous systems to the best of my knowledge, but there are not many reports regarding this aspect.
Do you have any plans for future work extending from this study?
As already mentioned above, we certainly will try to combine photooxidation and -reduction and we will use the functionalized membranes as coating for electrodes. The suggested “more active role of the membrane” is interesting and indeed the membrane could be more than a support for the catalyst.
The authors

Malte Hansen
Institute of Organic Chemistry, University of Regensburg, Germany
E-mail: malte.hansen@chemie.uni-regensburg.de

Dr Fei Li
State Key Laboratory of Fine Chemicals, DUT-KTH Joint Education and Research Centre on Molecular Devices, Dalian University of Technology (DUT), Dalian 116012, PR China
E-mail: lifei@dlut.edu.cn

Professor Licheng Sun
State Key Laboratory of Fine Chemicals, DUT-KTH Joint Education and Research Centre on Molecular Devices, Dalian University of Technology (DUT), Dalian 116012, PR China and Department of Chemistry, KTH Royal Institute of Technology, 10044 Stockholm, Sweden
E-mail:lichengs@kth.se

Professor Burkhard König
Institute of Organic Chemistry, University of Regensburg, Germany
E-mail: burkhard.koenig@ur.de
References
1. T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474-6502.
2. N. S. Lewis and D. G. Nocera, PNAS, 2006, 103, 15729-15735.
3. M. Hambourger, G. F. Moore, D. M. Kramer, D. Gust, A. L. Moore and T. A. Moore, Chem. Soc. Rev., 2008, 38, 25-35.
4. P. D. Frischmann, K. Mahata and F. Wurthner, Chem. Soc. Rev., 2013, 42, 1847-1870.
5. N. Kaveevivitchai, R. Chitta, R. Zong, M. El Ojaimi and R. P. Thummel, J. Am. Chem. Soc., 2012, 134, 10721-10724.
6. L. Kohler, N. Kaveevivitchai, R. Zong and R. P. Thummel, Inorg. Chem., 2013, 53, 912-921.
7. M. R. Norris, J. J. Concepcion, D. P. Harrison, R. A. Binstead, D. L. Ashford, Z. Fang, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2013, 135, 2080-2083.
8. F. Li, Y. Jiang, B. Zhang, F. Huang, Y. Gao and L. Sun, Angew. Chem. Int. Ed., 2012, 51, 2417-2420.
9. D. J. Wasylenko, R. D. Palmer and C. P. Berlinguette, Chem. Commun., 2013, 49, 218-227.
10. L. Duan, Y. Xu, M. Gorlov, L. Tong, S. Andersson and L. Sun, Chem. Eur. J., 2010, 16, 4659-4668.
11. A. Lewandowska-Andralojc and D. E. Polyansky, J. Phys. Chem. A, 2013, 117, 10311-10319.
12. L. Wang, L. Duan, L. Tong and L. Sun, J. Catal., 2013, 306, 129-132.
13. L. Duan, C. M. Araujo, M. S. G. Ahlquist and L. Sun, PNAS, 2012, 109, 15584-15588.
Additional information
Journal articles made easy are journal articles from a range of Royal Society of Chemistry journals that have been re-written into a standard, accessible format. They contain links to the associated Chemistry World article, ChemSpider entries, related journal articles, books and Learn Chemistry resources such as videos of techniques, and resources on theory and activities. They should facilitate students understanding of scientific journal articles and how to extract and interpret the information in them.



No comments yet