









Nuclear magnetic resonance (NMR) spectroscopy explores the electronic environment of atoms. A powerful technique useful for identifying the small to the very large
When some atoms are placed in a strong magnetic field, their nuclei behave like tiny bar magnets aligning themselves with the field.
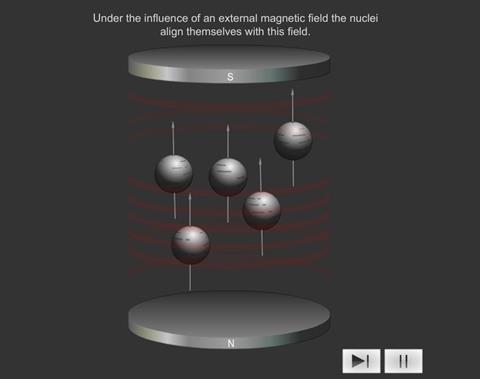
Electrons behave like this too, and for this reason both electrons and nuclei are said to possess “spin”, i.e. any spinning electric charge has an associated magnetic field.
Just as electrons with opposite spin pair up with each other, a similar thing happens with protons and neutrons in the nucleus. If a nucleus has an even number of protons and neutrons (e.g. 12 C), their magnetic fields cancel each other out and there is no overall magnetic field; however, if the number of protons and neutrons is odd (e.g.13 C and 1 H ), the nucleus has a magnetic field. If the substance is placed in an external magnetic field, the nuclear magnet lines up with the field, in the same way as a compass needle lines up with a magnetic field. The nuclear magnet can have two alignments, of low energy and high energy.
The basics - supplying energy
To make the nucleus change to the higher energy alignment, energy must be supplied.
The energy absorbed corresponds to radio frequencies. The precise frequency of energy depends on the environment of the nucleus, that is, on the other nuclei and electrons in its neighbourhood.
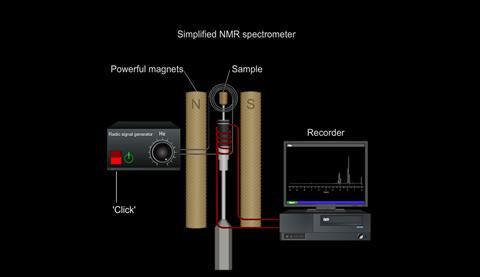
So, by placing the sample being examined in a strong magnetic field and measuring the frequencies of radiation it absorbs, information can be obtained about the environments of nuclei in the molecule.
Applications of NMR
NMR is one of the most powerful methods for analysis of chemical samples, biological compounds, medicines etc.
- Gives structural information on a range of different elements in molecules including hydrogen, carbon, phosphorus and many others.
- Is commonly used for structure determination of molecules in solution.
- Gives information regarding motion in molecules, structural flexibility and how they interact during chemical reactions.
- Can be used to determine shapes and structures of large complex molecules, such as how proteins fold, twist and coil.
- Can be used to evaluate the proportions of solid and liquid components in fatty foodstuffs such as margarines and low-fat spreads.
NMR is also used a lot in pharmaceutical sciences and medicine, for example:
- dynamic studies
- diagnosis of tissue abnormalities
- pH control in diabetics
- body scanning by the closely related techniques known as MRI (no side effects)
Downloads
Introducing a magnetic field
Simulation | Flash, Size 53.05 kbNuclear magnetic resonance spectrometer
Simulation | Flash, Size 13.94 kb
Nuclear magnetic resonance (NMR) spectroscopy
- 1
 Currently reading
Currently readingIntroduction
- 2







No comments yet