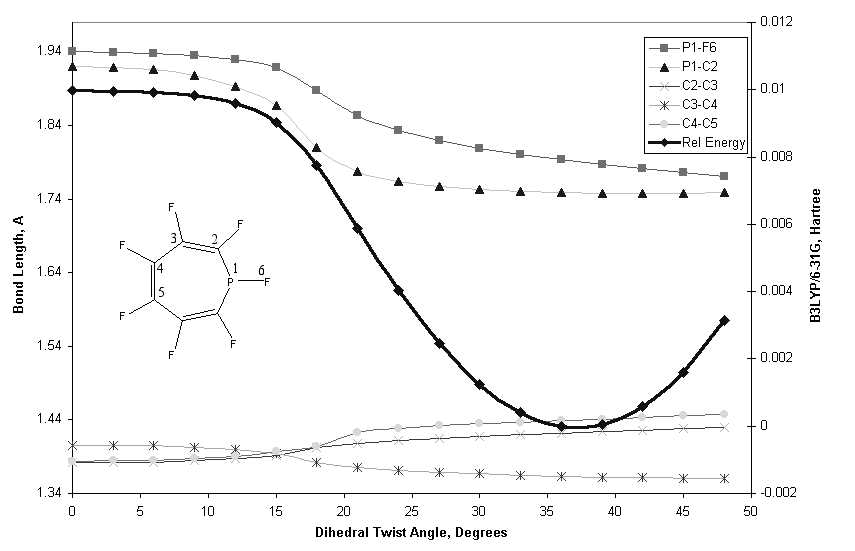
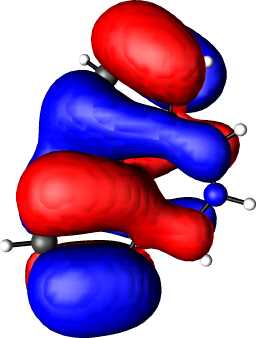
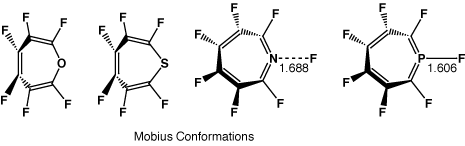
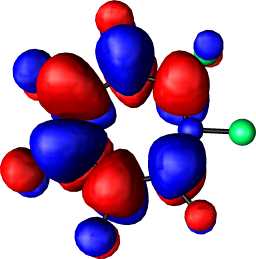
This journal is © The Royal Society of Chemistry 2002
| Table 1. Energies, kcal mol-1, relative to 2 or 4 (B3LYP/6-31G(d) in Hartree) and computed NICS Values (ppm). | ||||||
|---|---|---|---|---|---|---|
| System | Point Group | C=C configuration |
Negative Roots |
Dihedral angle C3-C4-C5-C6 |
Relative Energy (2 or 4 in parentheses) |
NICS(0) |
| 1a | Cs | cis,cis,cis | 0 | 0 | 2.6 (-902.7656) | -5.7 |
| 1a | C2 | cis,trans,cis | 0 | 94.8 | 36.3 | -10.5 |
| 1a | C2v | cis,cis,cis | 1a | 0.0 | 14.5 | +9.4 |
| 1b | Cs | cis,cis,cis | 0 | 0 | 4.7 (-1225.7418) | -8.0 |
| 1b | C2 | cis,trans,cis | 0 | 107.7 | 36.7 | -10.9 |
| 1b | C2v | cis,cis,cis | 1b | 0.0 | 21.3 | +7.3 |
| 1c | Cs | cis,cis,cis | 0 | 0 | -21.3 {-24.7}g (-982.0395) | -9.5 |
| 1c | C2 | cis,cis,cis (7c) | 0 | 32.4 | 4.9 {-1.1}g | -6.6 |
| 1c | C1 | cis,trans,cis | 0 | 98.1 | 23.9 | -10.7 |
| 1c | C2v | cis,cis,cis (5c) | 2c | 0.0 | 14.1 | 17.5 |
| 1c | C2v | cis,cis,cis (8c) | 2d | 0.0 | 27.8 {4.2}g | - |
| 1d | Cs | cis,cis,cis | 0 | 0 | -11.4 {-9.8}g (-1268.7453) | -7.9 |
| 1d | C2 | cis,cis,cis (7d) | 0 | 43.9 | 46.0 {47.7}g | -10.9 |
| 1d | C1 | cis,trans,cis | 0 | 111.0 | 23.0 | -6.5 |
| 1d | C2v | cis,cis,cis (6d) | 3e | 0.0 | 81.9 | 53.4 |
| 1d | C2v | cis,cis,cis (8d) | 3f | 0.0 | 67.0 {63.1}g | -8.5 |
| 3a | C2 | cis,cis,cis | 0 | 39.4 | 40.5 (-614.7114) | 26.5, -1.1h |
| 3b | C2 | cis,cis,cis | 0 | 42.7 | 25.5 (-799.1745) | 16.5, -3.1h |
| 3c | C2 | cis,cis,cis | 0 | 53.8 | 46.2 (-937.7039) | 14.9, -4.0h |
| 3d | C2 | cis,cis,cis | 0 | 41.2 | 19.3 (-594.8438) | 20.8,-1.0h |
| 3e | C2 | cis,cis,cis | 0 | 58.4 | 52.6 (-881.4595) | 8.2, -6.2h |
| 3f | C2 | cis,cis,cis | 0 | 60.2 | 34.6 (-1065.9222) | 4.0, -7.7h |

| 5c | Symmetry | 7c | 8c | Symmetry | |
|---|---|---|---|---|---|
 |
a2 | a | 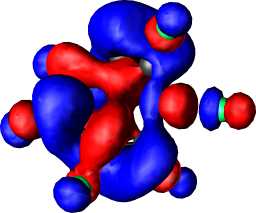 |
 |
b1 |
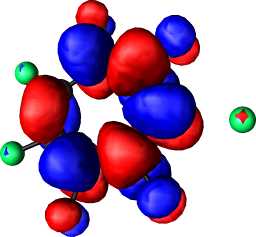
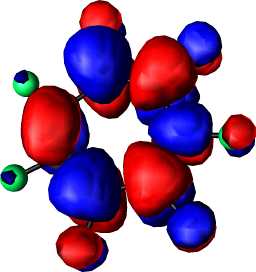 |
b1 | a | 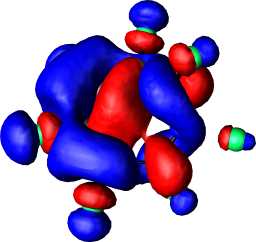 |
 |
a2 |
 |
a1 | b |  |
 |
a1 |
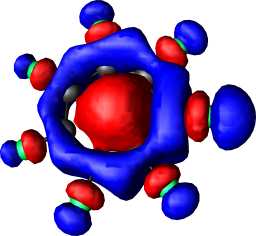 |
a1 | a | 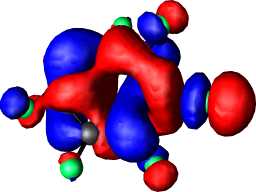 |
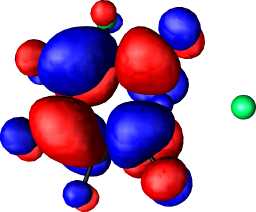 |
a2 |
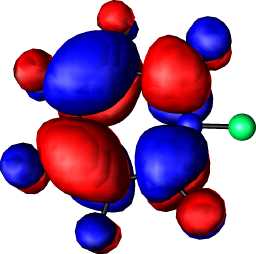 |
a2 | b | 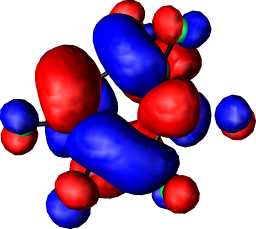 |
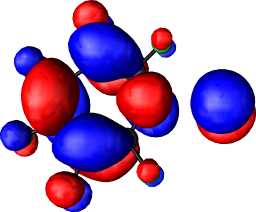 |
b1 |
| HOMO-LUMO gap | |||||
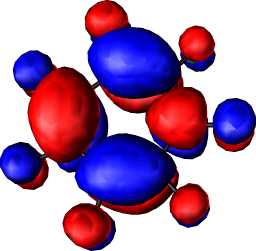 |
b1 | a | 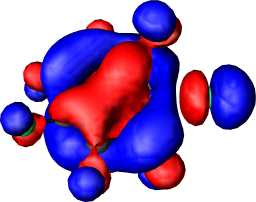 |
 |
b1 |
 |
a2 | a |  |
 |
a1 |
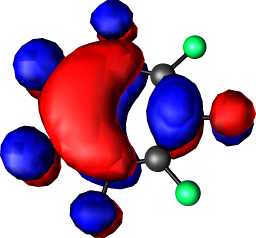 |
b1 | b | 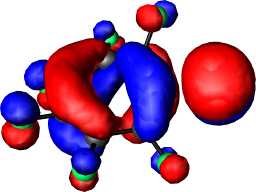 |
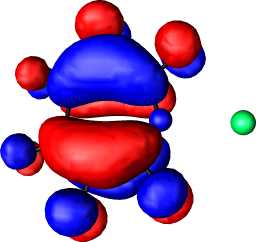 |
a2 |
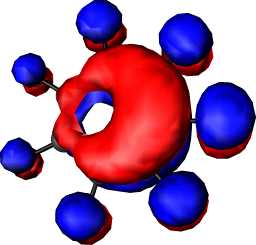 |
b1 | b |  |
 |
a1 |
 |
b1 | b |  |
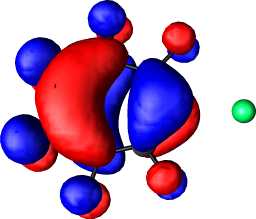 |
b1 |
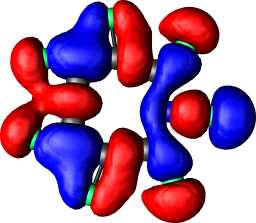 |
a1 | a | 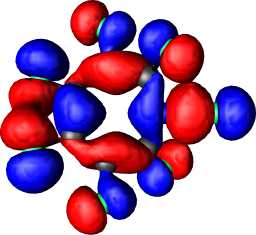 |
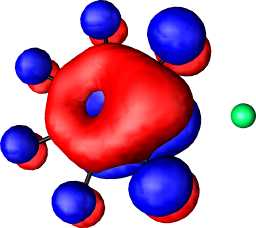 |
b1 |

| 5d | Symmetry | 6d | 7d | Symmetry | |
|---|---|---|---|---|---|
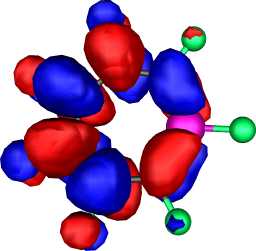 |
a2 | b | 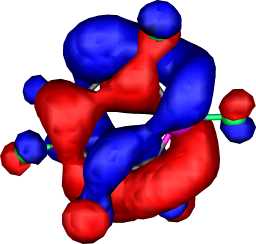 |
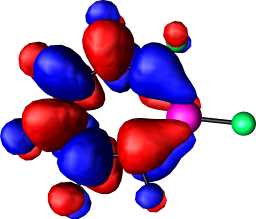 |
a2 |
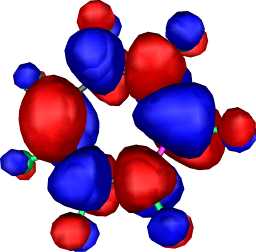 |
b1 | a | 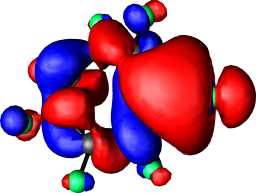 |
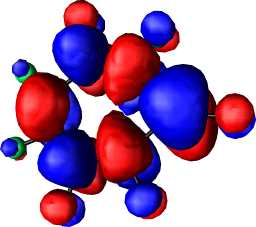 |
b1 |
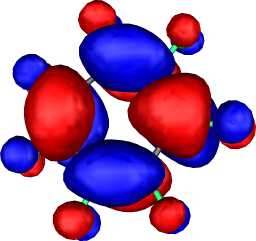 |
b1 | b | 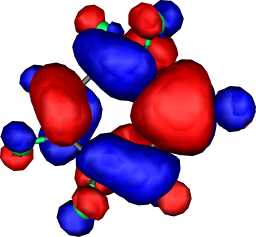 |
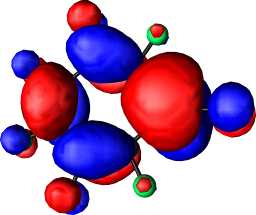 |
b1 |
| HOMO-LUMO gap | |||||
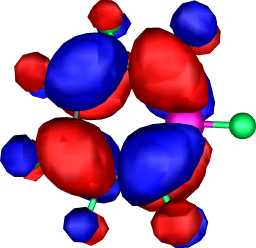 |
a2 | a | 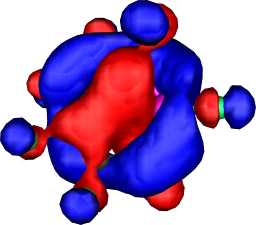 |
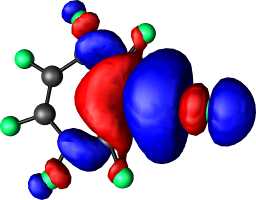 |
a1 |
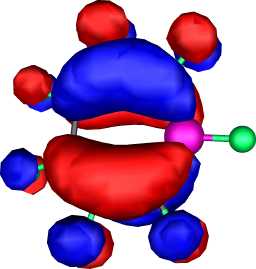 |
a2 | a | 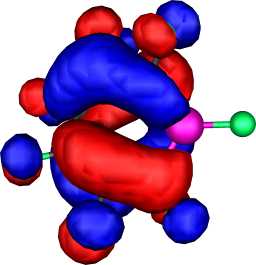 |
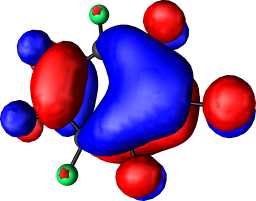 |
b1 |
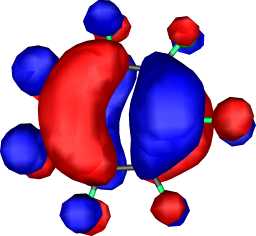 |
b1 | b | 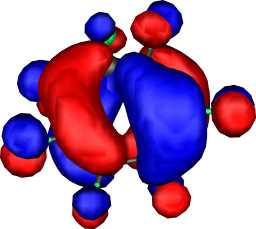 |
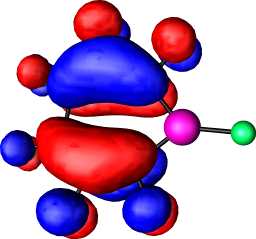 |
a2 |
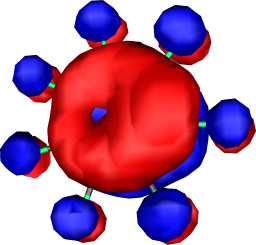 |
b1 | b | 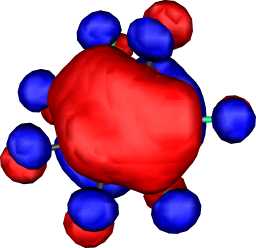 |
 |
b1 |
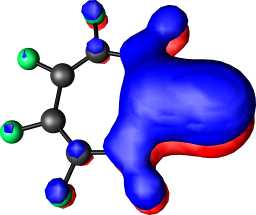 |
b1 | b | 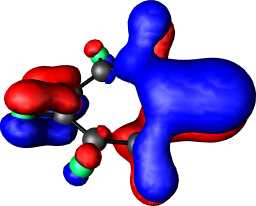 |
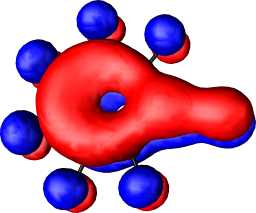 |
b1 |