 2
instead of , where = 0.0068 eV
in Ar and 0.0072 eV in Kr.
2
instead of , where = 0.0068 eV
in Ar and 0.0072 eV in Kr.| Phys. Chem. Chem. Phys., 2004, 6 | |
Additions and corrections A potential energy surface and a trajectory study of photodynamics and strong-field alignment of ClF molecule in rare gas (Ar,Kr) solids |
Toni Kiljunen, Matias Bargheer, Markus Gühr and Nikolaus Schwentner
Phys. Chem. Chem. Phys., 2004, 6, 2185 (DOI: 10.1039/b315149b). Amendment published 6th August 2004
In the computation of the potential energy surfaces, in the range
of 2.45 < r < 5.25 Å for F–Ar and 2.46375 < r < 5.475 Å for
F–Kr, the Morse functional part was energy-scaled mistakenly by
2
instead of , where = 0.0068 eV
in Ar and 0.0072 eV in Kr.
The trajectory calculation, and thus the main body of the article, remains unaffected since the potential gradients are free from this error. The conclusions were mainly drawn from the dynamics results, and thereby remain valid. Also for the potential energy plots in Figs. 5, 11 and 15, the difference remains within the line width of the curves for stretching into the tetra-atomic window and at the maximal error region (triatomic barrier) it stays below 0.1 eV.
The dominant change appears in Fig. 2 for Ar which is therefore shown here. The triatomic window is now the most repulsive direction in agreement with the dynamical result. The tetra-atomic window maintains its preference as the minimum energy orientation for the molecule. In Kr, the qualitative result for energies (nearest neighbour < triatomic < tetra-atomic window) is not changed—only the representative numbers, which are now given in the new Table 1, below. Consequently, the potentials obtained after the thermalization or the geometry optimization stages, shown in Figs. 3 and 6 of the article, become modified. We present corrected numbers for the optimization case in Table 1, and note that whereas in Ar the triatomic window has a more pronounced repulsive energy surface, the potential shape representing the instantaneous barriers around the solvated molecule remain in Kr the same as before.
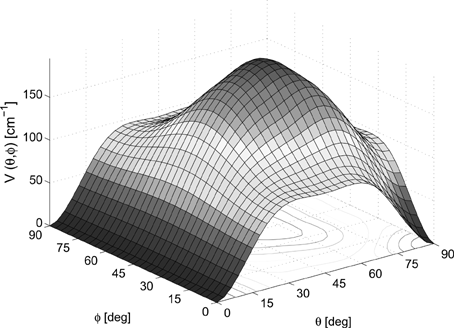
Fig. 2 The corrected potential energy landscape for rotating the ClF in a perfect Ar lattice. The crystal size corresponds to the 5 K case where the lattice constant is 5.2238 Å.
Table 1| Ar |
Kr |
||||||
| T/K | 3w | 4w | nn | 3w | 4w | nn | nn  nn nn |
| 5 | 195 | 0 | 93 | 135 | 156 | 0 | 104 |
| 19 | 181 | 0 | 95 | 135 | 156 | 0 | 105 |
T | 148 | 0 | 93 | 134 | 154 | 0 | 106 |
| After the geometry optimization | |||||||
| 5 | 289 | 0 | 184 | 157 | 238 | 0 | 123 |
The Royal Society of Chemistry apologises for these errors and any consequent inconvenience to authors and readers.