Fighting cystic fibrosis
Our ChemSci Pick of the Week marks an important step towards a new therapy addressing the fundamental cause of cystic fibrosis.
Cystic fibrosis is one of the UK's most common life-threatening hereditary diseases. Around 11,000 people in the UK live with the condition, and one in 25 people are genetic carriers. While treatments to alleviate its symptoms are improving, the average life expectancy of someone with cystic fibrosis is still only 38 years.
An international team of researchers have published a paper in Chemical Science which marks an important step towards a new therapy addressing the fundamental cause of the disease.
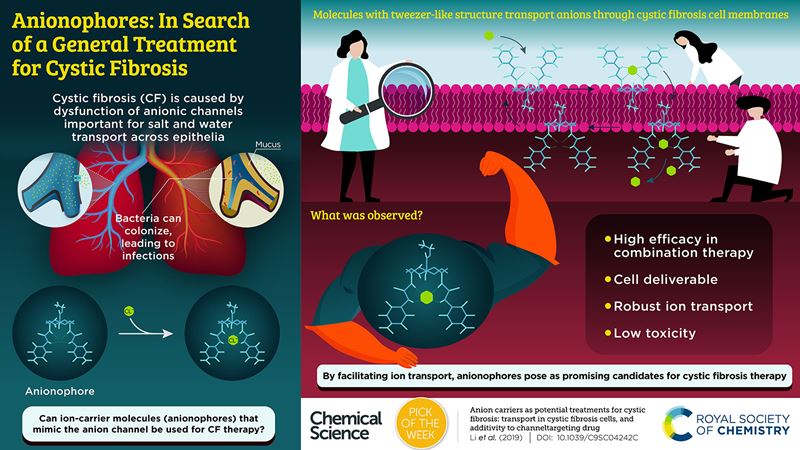
Corresponding author Professor Anthony Davis FRSC from the University of Bristol, UK, explains that cystic fibrosis occurs when someone possesses two recessive copies of a certain gene. This means that they lack a key molecular component at the surface of cells in their lungs, and elsewhere in their body.
Professor Davis said: "The molecule is called the Cystic Fibrosis Conductance Regulator (CFTR), and its job is to allow the passage of negatively charged species – anions – across the cell membrane. If CFTR is absent, it affects the composition of the mucus in the lungs, which becomes thick and sticky, and liable to infection."
The team’s solution involves designing synthetic "anion carrier" molecules, which could be delivered to a patient’s cell membrane to do the same job as the missing CFTR. In this paper they report a new method for testing these molecules in cells, as well as several new active compounds.
"We also show that the compounds need not be toxic, which is an important issue, and that their activity can supplement the effects of recently introduced drugs, which help some CF patients through a different mechanism," said Professor Davis.
"There’s still a long way to go, but if the research proceeds as hoped it might lead to a genuinely effective and general treatment for CF patients over this timescale."
This article is free to read in our open access, flagship journal Chemical Science: Hongyu Li et al. Chem. Sci., 2019, Advance Article. DOI: 10.1039/c9sc04242c. You can access our 2019 ChemSci Picks in this article collection.