Additions and corrections
Rapid polymorph screening on milligram
quantities of pharmaceutical material using phonon-mode Raman spectroscopy
Sarah
Al-Dulaimi, Adeyinka Aina and Jonathan Burley
CrystEngComm, 2010, 12, 1038 (DOI: 10.1039/b921114b). Amendment published 24th June 2010
In a recent publication we outlined the use of low-wavenumber Raman spectroscopy for rapid screening of polymorphism in pharmaceutical materials.1 We presented data collected on three model systems to illustrate the efficacy of our approach. One of the systems employed was paracetamol, which was selected as it is a well-characterised model system. Because of the well-characterised nature of this model, we did not undertake a thorough and rigorous analysis of our data, but instead based our interpretation on previous work undertaken by one of the authors (JB), published earlier,2 and supported by a body of other work.3,4
Further, more detailed experiments have indicated that this interpretation, which at the time appeared reasonable, was not valid. As a result the assignments presented in our recent work in CrystEngComm for the Raman spectra of the different solid forms of paracetamol are incorrect. This affects Fig. 1(a and b) and also affects our discussion of the in-situ crystallisation based on data presented in Fig. 2a. The main thrust of our paper is unaffected, and the results and discussion of the two other systems presented in the work remain valid.
In Fig. 1 a and 1b, the spectra labelled I and II in fact both correspond to form I, and that labelled III in fact corresponds to form II. A corrected version of Fig. 1 is given below, changes have been made to panes a and b only.
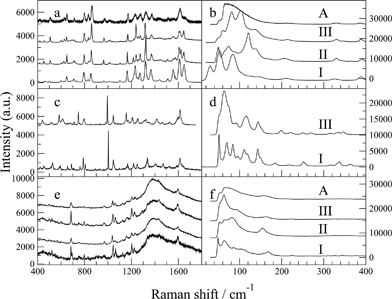
Fig. 1 Raman spectra of the solid forms of: paracetamol (a, b); FFA (c, d); imipramine hydrochloride (e, f). A = amorphous.
Likewise, in Fig. 2a of our original publication, the spectra were incorrectly ascribed to a series of transformations from amorphous to form III to form II. With the benefit of a thorough review of the data and more detailed experiments, it has become clear that these data in fact correspond to a transformation sequence amorphous to form II (not III), then to form I (not II).
Our error in assuming reproducibility of the crystallisation pathway of amorphous paracetamol was compounded by the fact that we collected spectra at 10 °C intervals, which meant that unambiguous identification of polymorphic forms via their melting points was not possible. The collection of data every 10 °C was due to technical constraints in place at the time of the original experiments which have since been resolved. To avoid any similar mistake in future, we have updated our screening protocol to ensure that spectra are collected every 1 °C in future, and are employing statistical tests rather than visual observation to differentiate between spectra. We also hope that the recent development of density-functional theory to a level whereby confident peak assignments can be made for solid-state Raman spectra5 will prove invaluable in future work. We apologise wholeheartedly for any confusion caused to the readership of CrystEngComm.
Acknowledgements
We thank Dr Jagadeesh Nanubolu for bringing this error to our attention.
References
1. S. Al-Dulaimi, A. Aina and J. Burley, CrystEngComm, 2010, 12, 1038–1040.
2. J. C. Burley, M. J. Duer, R. S. Stein, and R. M. Vrcelj, Eur. J. Pharm. Sci., 2007, 31, 271–276.
3. P. Martino, P. Conflant, M. Drache, J. Huvenne and A. Guyot-Hermann, J. Thermal Anal., 1997, 48, 447–458.
4. S. Qi, P. Avalle, R. Saklatvala and D. Q. Craig, Eur. J. Pharm. Biopharm., 2008, 69, 364–371.
5. P. U. Jepsen and S. J. Clark, Chem. Phys. Lett., 2007, 442, 275–280.
The Royal Society of Chemistry apologises for this error and any consequent inconvenience to authors and readers.