- Home
- I am a …
- Resources
- Collections
- Post-lockdown teaching support
- Remote teaching support
- Starters for ten
- Screen experiments
- Assessment for learning
- Microscale chemistry
- Faces of chemistry
- Classic chemistry experiments
- Nuffield practical collection
- Anecdotes for chemistry teachers
- More …
- Literacy in science teaching
- Climate change and sustainability
- Alchemy
- On this day in chemistry
- Global experiments
- PhET interactive simulations
- Chemistry vignettes
- Context and problem based learning
- Journal of the month
- Chemistry and art
- Classic chemistry demonstrations
- In search of solutions
- In search of more solutions
- Creative problem-solving in chemistry
- Solar spark
- Chemistry for non-specialists
- Health and safety in higher education
- Analytical chemistry introductions
- Exhibition chemistry
- Introductory maths for higher education
- Commercial skills for chemists
- Kitchen chemistry
- Journals how to guides
- Chemistry in health
- Chemistry in sport
- Chemistry in your cupboard
- Chocolate chemistry
- Adnoddau addysgu cemeg Cymraeg
- The chemistry of fireworks
- Festive chemistry
- Collections


- Education in Chemistry
- Teach Chemistry
- Events
- Teacher PD
- Enrichment
- Our work
- More from navigation items








Analytical chemistry introductions
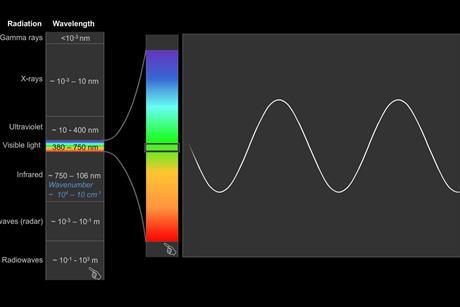
Introduction to spectroscopy
Get back to basics with this primer on the principles of spectroscopic techniques, including infrared (IR), ultraviolet-visible (UV-vis) and nuclear magnetic resonance (NMR). To make it even easier, each technique has clear explanations and descriptions supported by animations.

Infrared (IR) spectroscopy
Absorption of infrared radiation brings about changes in molecular vibrations within molecules and ‘measurements’ of the ways in which bonds vibrate gives rise to infrared spectroscopy. Atom size, bond length and bond strength vary in molecules and so the frequency at which a particular bond absorbs infrared radiation will be different over a range of bonds and modes of vibration.

Ultraviolet–visible (UV-vis) spectroscopy
Learn how UV-visible radiation can be used to shed light on chemical identification and how our senses percept colour. From the theory behind molecular orbitals and electronic transitions to the application of this technique with relatable examples. Includes examples and interactive simulations to aid understanding.

Nuclear magnetic resonance (NMR) spectroscopy
Discover how nuclear magnetic resonance (NMR) spectroscopy works, with this series of topics breaking down the fundamental theory. Covering the electronic environment of atoms right up to demonstrating the practical identification of molecules. Includes examples and interactive simulations to aid understanding.
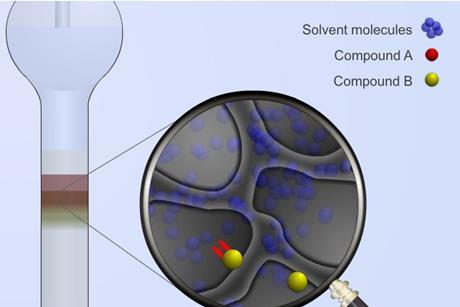
Chromatography
Chromatography covers a broad range of physical methods used to separate and/or analyse complex mixtures. It can be preparative or analytical and has a wide range of applications.
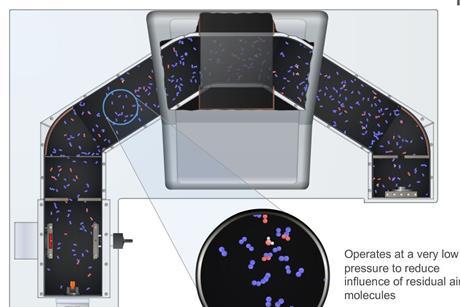
Mass spectrometry (MS)
Mass spectrometry is a powerful technique in the modern analytic laboratory. Learn the fundamental theory behind the operation of a mass spectrometer.